-
-
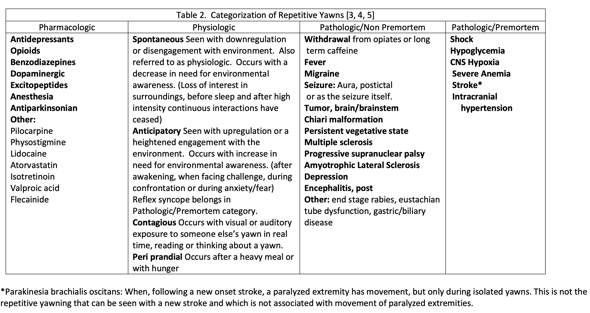 -
- Premortem yawning is absent from almost all
major medical textbooks but has been associated
for more than 2,000 years with impending death
from acute infections and hemorrhage and was
commonly known during the Bubonic Plague
pandemic. [1,2] Medical reports of
premortem yawning identify multiple causes of
shock including vasovagal reflex, severe
hypoxia/anemia/hypoglycemia, stroke, and
intracranial hypertension. [3,4,5]
-
- Six emergency department patients
experienced new onset, repetitive yawning,
coincident with new onset hypotension, and in
four of those cases, also a depressed heartrate.
In all cases, the yawns recurred every two to
five minutes, were high volume, wide open mouth,
and irrepressible. The autonomic nervous system
(ANS) usually directs actions that ensure
survival of the body in which it is contained,
yet in Phase II shock, the ANS may suddenly
promote rapid death. [6] Yawning, can
herald such a change.
-
- The most probable physiologic explanation
for a shock/yawn pathway follows: A decrease of
CNS ATP production causes excess CNS glutamate
within the paraventricular nucleus (PVN),
followed by glutamate activation of yawning,
hypotension, and bradycardia. Decreased CNS ATP
results from multiple causes of CNS tissue
hypoxia but also occurs with adequate
oxygenation in the presence of cyanide, a poison
of the mitochondrial cytochrome system.
[7]
-
- CNS ATP production is vulnerable to failure
considering that a neuronal cell consumes more
than ten million ATP molecules per second, yet
CNS neuronal ATP production is on-site,
aerobic-only, with a mere ten seconds of
reserve. One protective mechanism for ATP
production is ongoing autoregulation of cerebral
blood flow (CBF). When CNS hypoxia or decreased
perfusion occurs, ongoing selective rationing of
blood flow throughout the brain, changes within
seconds [8]. This activity begins before
vital signs change.[9] When cerebral
blood flow is reduced by fifty percent,
autoregulation capability becomes inadequate,
and systemic hypotension occurs. [10]
This is likely from CNS hypoxia initiating a
yawn/shock pathway. Clinically, elevation of a
patient's head by ninety degrees decreases
cerebral perfusion pressures by 30
mmHg.[11]
-
- Glutamate is one thousand to ten
thousand-fold more prevalent than other CNS
neurotransmitters, is excitatory, and at high
concentrations is a neuronal excitotoxin.
Following a CNS action potential, glutamate is
normally cleared from the synaptic cleft by
transporters that require ATP.[12] As
CNS ATP production fails, glutamate accumulates
in effected, extracellular, CNS tissue
[13] including the PVN. The PVN contains
a yawn modulation center [14] and a
cardiovascular modulation center, one more
likely to be depressant than
stimulatory.[15] Glutamate excess within
the PVN causes yawning, [16 ]
bradycardia[17 ] and potentiates
hypotension.[18]
-
- The culprit causes of shock in these six
cases are, traumatic hemorrhage, sepsis, ST
elevation myocardial infarction (STEMI), non-ST
elevation myocardial infarction (NSTEMI), severe
anemia from heavy menstrual bleeding (HMB) and
vasovagal asystole (VVA). [Table 1] One
or more additional circulatory stressors
occurred in four of the cases and include
profound fluid loss through diaphoresis,
nitroglycerin induced preload reduction, other
medication, iatrogenic hemodilution, and rapid
elevation of the head of the bed.
-
- In all cases, when yawning was first
observed, depression of either blood pressure or
heart rate was present. Time from last known
normal blood pressure to yawning, varied from
one to ten minutes. Continuous peripheral pulse
oxygenation (PPO) exceeded 95% except in the
case of nonviable hemoglobin. Yawning ceased
during recovery when vital signs approached
normal. In the cases of traumatic hemorrhage and
sepsis, yawning resolved with volume infusion
and supine position/passive leg raise. In the
cases of STEMI and NSTEMI, additional management
included discontinuation of nitroglycerin, and
in the latter case, intravenous atropine. The
cases of HMB and WA were the most severe, and
head elevation was rapidly followed by
cardiovascular collapse and repetitive
yawning.
-
- The case of HMB with iatrogenic
hemodilution, had sudden, profound, unwavering
bradycardia and hypotension, unresponsive to
oxygen, supine position, and a total of 2 mg of
intravenous atropine. However, at the precise
time, a viable hemoglobin level was restored,
the monitored heart rate and blood pressure
abruptly corrected in less than one minute as
CNS oxygenation and ATP production were restored
with clearing of CNS intercellular
glutamate.
-
- VVA occurs at the point that cardiac output
is maximized with progressive circulatory
stress. [19] In this case, head
elevation caused hypoperfusion of the CNS,
exceeding the ability of cerebrovascular
autoregulation. CNS hypoxia followed, with more
than expected glutamate surge because of heavy
neuronal traffic between the amygdala and PVN
from the patient's profound panic.
-
- Although yawning is usually benign, when
repetitive yawning occurs in the emergency
patient, it may warn of a premortem or
permanently disabling condition. Assessment
includes new vital signs, point-of-care glucose,
an exam with concern for shock, severe
hypoglycemia/hypoxia/anemia, stroke, or
increased intracranial pressure.
-
-
- References
- [1] Walusinski 0. Historical
perspectives. In: Walusinski O, editor. The
mystery of yawning in physiology and disease.
Front Neurol Neurosci. Basel, Karger,2010;28, p
1-21.
- [2] Walusinski O. Popular knowledge
and beliefs. In: Walusinski 0 editor. The
mystery of yawning in physiology and disease.
Front Neur Neurosci. Basel, Karger, 2010;28, p
22-25.
- [3] Krestel H, Bassetti C,
Walusinski O. Yawning- Its anatomy, chemistry,
role and pathological considerations. Progress
in Neurobiology. 2018 Feb;161:61-78.
- [4] Walusinski O. Associated
diseases. In: Walusinski 0, volume editor. The
mystery of yawning in physiology and disease,
Front Neurol Neurosci:Basel Karger;2010, p.
140-55.
- [5] [13] Cronin TG Jr.
Yawning: an early manifestation of vasovagal
reflex. AJR Am J Roentgenol 1988
Jan;150(l):209.
- [6] Schadt JC, Ludbrookj.
Hemodynamic and neurohumeral responses to acute
hypovolemia in conscious mammals. Am J Physiol
1991;260:H305-18.
- [7] Morocco AP. Cyanides. Crit Care
Clin 2005;21:691-705.
- [8] Hall JE, Guyton AC. Cerebral
blood flow, cerebral spinal fluid, and brain
metabolism. In: Hall JE, Guyton AC, (editors).
Guyton and Hall textbook of medical physiology,
Philadelphia PA, Saunders, Twelfth Edition, p
743-50.
- [9] Sufladowicz E, Maniewski R,
Kozluk E, Zbiec A, Nosek A, Walczak F. Near
infrared spectroscopy in evaluation of cerebral
oxygenation during vasovagal syncope. Physiol
Meas. 2004;25:823-36.
- [10] Cheng R, Shang Y, Wang S, Evans
JM, Rayapati A, Randall DC, et al: Near-infrared
diffuse optical monitoring of cerebral blood
flow and oxygenation for the prediction of
vasovagal syncope. J Biomed Opt
2014;19(1):17001.
- [11] Rosner MJ, Coley IB. J
Neurosurg. 1986;65(5):636.
- [12] Baur D, Robinson M.
Glutamatergic neurotransmission In: Robertson D,
Biaggioni l,9G, Low PA, Paton JFR, editors.
Primer on the autonomic nervous system Third
Edition, Academic Press P 103-7.
- [13] Foo K, Blumenthal L, Man H.
Regulation of neuronal bioenergy homeostasis by
glutamate. Neurochem Internat.
2012,61:389-396.
- [14] Collins GT, Eguibar JR. Chap 11
Neuropharmacology of Yawning in Walusinski O
(ed), The Mystery of Yawning in Physiology and
Disease. Front Neur Neurosci. Basel, Karger,
2010;28:90- 106.
- [15] Hall JE, Guyton AC. Nervous
regulation of the circulation, and rapid control
of arterial pressure. In: Hall JE, Guyton AC,
(editors). Guyton and Hall textbook of medical
physiology, Philadelphia PA, Saunders, Twelfth
Edition, p 203.
- [16] Kita E, Sato-Suzuki I, Oguri M,
Arita H. Yawning responses induced by local
hypoxia in the paraventricular nucleus of the
rat. Behav Brain Res. 2000, Dec
20;117(l-2):119-26.
- [17] Crestani CC, Alves FH, Busnardo
C, Resstel LB, Correa FM. N-Methyl-D-aspartate
glutamate receptors in the hypothalamic
paraventricular nucleus modulate cardiac
component of the baroreflex in unanesthetized
rats. Neurosci Res 2010;67:317-26.
- [18] Busnardo C, Crestani CC,
Fassini A, Resstel LB, Correa FM. NMDA and
non-NMDA glutamate receptors in the
paraventricular nucleus of the hypothalamus
modulate different stages of hemorrhage-evoked
cardiovascular responses in rats. Neuroscience.
2016 Apr 21;320:149-59.
- [19] [40] Jardine DL, Ikram
H, Crozier IG. Autonomic control of asystolic
vasovagal syncope. Heart. 1996;
75(5):528-30.
-
-
-
- Le bâillement pré-mortem est
absent de presque tous les principaux manuels
médicaux, mais il est associé
depuis plus de 2000 ans à la mort
imminente d'infections aiguës et
d'hémorragies et était
communément connu pendant la
pandémie de peste bubonique.
[1,2] Les rapports médicaux de
bâillements pré-mortem identifient
de multiples causes de choc, notamment le
réflexe vasovagal, une hypoxie /
anémie / hypoglycémie
sévère, un accident vasculaire
cérébral et une hypertension
intracrânienne. [3,4,5]
-
- Chez six patients d'un service d'urgence
sont apparus des bâillements
répétitifs, coïncidant avec
l'installation d'une hypotension, et dans quatre
de ces cas, également un rythme cardiaque
ralenti. Dans tous les cas, les
bâillements se reproduisaient toutes les
deux à cinq minutes, de façon
répétée, bouche grande
ouverte et irrépressibles. Le
système nerveux autonome (SNA) dirige
généralement des actions qui
assurent la survie de l'individu, mais en cas de
choc de phase II, le SNA peut soudainement
favoriser une mort rapide. [6] Le
bâillement peut annoncer un tel
changement.
-
- L'explication physiologique la plus probable
de l'état de choc / bâillement est
la suivante : Une diminution de la production
d'ATP du SNC provoque un excès de
glutamate du SNC dans le noyau paraventriculaire
(PVN), suivi d'une activation par le glutamate
du bâillement, de l'hypotension et de la
bradycardie. La diminution de l'ATP du SNC
résulte de multiples causes d'hypoxie des
tissus du SNC, mais se produit également
avec une oxygénation adéquate en
présence de cyanure, un poison du
système cytochromique mitochondrial.
[7]
-
- La production d'ATP du SNC est
vulnérable étant donné
qu'une cellule neuronale consomme plus de dix
millions de molécules d'ATP par seconde,
sachant que la production d'ATP neuronal du SNC
est locale, aérobie uniquement, avec
seulement dix secondes de réserve. Un
mécanisme de protection pour la
production d'ATP est l'autorégulation
continue du flux sanguin cérébral
(CBF). En cas d'hypoxie du SNC ou de diminution
de la perfusion, le rationnement sélectif
continu du flux sanguin dans le cerveau change
en quelques secondes [8]. Cette
activité commence avant le changement des
signes vitaux [9]. Lorsque le
débit sanguin cérébral est
réduit de cinquante pour cent, la
capacité d'autorégulation devient
insuffisante et une hypotension
systémique se produit. [10] Ceci
est probablement dû à l'hypoxie du
SNC qui a initié une voie de
bâillement / choc. Cliniquement,
l'élévation de la tête d'un
patient de quatre-vingt-dix degrés
diminue les pressions de perfusion
cérébrale de 30 mmHg.
[11]
-
- Le glutamate est mille à dix mille
fois plus répandu que les autres
neurotransmetteurs du SNC, est excitateur et,
à des concentrations
élevées, est une excitotoxine
neuronale. Après un potentiel d'action du
SNC, le glutamate est normalement
éliminé de la fente synaptique par
les transporteurs qui nécessitent de
l'ATP. [12] Comme la production d'ATP du
SNC échoue, le glutamate s'accumule dans
les tissus CNS extracellulaires affectés
[13], y compris le PVN. Le PVN contient
un centre de modulation du bâillement
[14] et un centre de modulation
cardiovasculaire, un plus susceptible
d'être déprimé que
stimulé. [15] L'excès de
glutamate dans le PVN provoque un
bâillement [16], une bradycardie
[17] et potentialise l'hypotension
[18].
-
- Les causes coupables de choc dans ces six
cas sont l'hémorragie traumatique, la
septicémie, l'infarctus du myocarde avec
élévation du segment ST (STEMI),
l'infarctus du myocarde avec
élévation non ST (NSTEMI),
l'anémie sévère due
à des saignements menstruels abondants
(HMB) et l'asystole vasovagale (VVA). Un ou
plusieurs facteurs de stress circulatoires
supplémentaires sont survenus dans quatre
des cas et comprennent une perte de liquide
profonde par diaphorèse, une
réduction de la précharge induite
par la nitroglycérine, d'autres
médicaments, une hémodilution
iatrogène et une élévation
rapide de la tête du lit.
-
- Dans tous les cas, lorsque le
bâillement a été
observé pour la première fois, une
dépression de la pression
artérielle ou de la fréquence
cardiaque était présente. Le temps
écoulé entre la dernière
pression artérielle normale connue et le
bâillement variait de une à dix
minutes. L'oxygénation pulsée
périphérique continue (PPO) a
dépassé 95% sauf dans le cas de
l'hémoglobine non viable. Le
bâillement a cessé pendant le
rétablissement lorsque les signes vitaux
se sont rapprochés de la normale. Dans
les cas d'hémorragie traumatique et de
septicémie, le bâillement s'est
résolu avec une perfusion volumique et
une position couchée sur le dos / une
élévation passive de la jambe.
Dans les cas de STEMI et NSTEMI, la prise en
charge supplémentaire comprenait
l'arrêt de la nitroglycérine et,
dans ce dernier cas, l'atropine intraveineuse.
Les cas de HMB et WA étaient les plus
graves et l'élévation de la
tête était rapidement suivie d'un
effondrement cardiovasculaire et de
bâillements répétitifs.
-
- Le cas de HMB avec hémodilution
iatrogène a eu une bradycardie et une
hypotension soudaines, profondes et
irrécupérables, ne
répondant pas à l'oxygène,
en position couchée et un total de 2 mg
d'atropine intraveineuse. Cependant, à
l'heure précise, un taux
d'hémoglobine viable a été
restauré, la fréquence cardiaque
et la pression artérielle
surveillées ont été
brusquement corrigées en moins d'une
minute, car l'oxygénation du SNC et la
production d'ATP ont été
restaurées avec l'élimination du
glutamate intercellulaire du SNC.
-
- La VVA survient au point où le
débit cardiaque est maximisé avec
un stress circulatoire progressif. [19]
Dans ce cas, l'élévation de la
tête a provoqué une hypoperfusion
du SNC, dépassant la capacité
d'autorégulation
cérébrovasculaire. Une hypoxie du
SNC a suivi, avec une augmentation du glutamate
plus que prévu en raison de
l'activité neuronale intense entre
l'amygdale et le PVN à cause de la
panique profonde du patient.
-
- Bien que le bâillement soit
généralement bénin, lorsque
les bâillements répétitifs
apparaissent chez un patient aux urgences, il
peut avertir d'une condition prémortem ou
d'une incapacité permanente.
L'évaluation comprend de nouveaux signes
vitaux, la glycémie, un examen avec
préoccupation pour le choc, une
hypoglycémie / hypoxie / anémie
sévère, un accident vasculaire
cérébral ou une augmentation de la
pression intracrânienne.
|


