Lymphomatoid
Granulomatosis After Childhood Acute
Lymphoblastic Leukemia: Report of Effective
Therapy
Christopher L. Moertel, Bonnie
Carlson-Green,
Jan Watterson, Susan C. Simonton
Departments of
Hematology/Oncology, Child and
Family Services, and Pathology, Children's
Hospitals and Clinics, St Paul,
Minnesota.
Lymphomatoid granulomatosis, a rare
condition in children, affects the lungs
primarily but may have significant
extrapulmonary manifestations, especially in the
central nervous system. We report a case of
lymphomatoid granulomatosis with onset after the
completion of chemotherapy for childhood acute
lymphoblastic leukemia. Two months after
treatment ended, the 7-year-old girl developed
splenomegaly, cervical adenopathy, and bilateral
interstitial pulmonary infiltrates. She improved
on cefotaxime but experienced a seizure
1 month later.
A computed tomography scan of the head was
normal, but her pulmonary infiltrates had become
nodular. A computed tomography-guided biopsy of
1 of the nodules revealed cellular
interstitial pneumonitis. One month later, she
had persistent pulmonary infiltrates, marked
splenomegaly, and new seizures. Magnetic
resonance imaging of the head revealed cerebral
nodules. Itraconazole was begun, and the
pulmonary infiltrates resolved.
Five months after her initial symptoms, she
developed tonic pupil and a decreased level of
consciousness. Dexamethasone was initiated.
Needle biopsies of the brain were carried out,
yielding the diagnosis of severe chronic
inflammatory changes focally consistent with
granuloma. The child redeveloped splenomegaly
and fever, and then suffered an acute
decompensation with hypoxemia, tachypnea,
splenomegaly, and cardiac gallop.
Open-lung biopsy revealed lymphomatoid
granulomatosis. Lymphoma-directed therapy was
initiated, and the patient had complete
resolution of pulmonary and cerebral nodules
5 months later. No intrathecal chemotherapy
was administered, and radiation therapy was not
necessary. Neuropsychological testing obtained
after completion of therapy revealed an
improvement in attention, coordination, and fine
motor speed over time. She is now in good health
and attending school.
A 5-year-old girl was diagnosed with ALL in
September 1992. No central nervous system
disease was detected. Lymphoblasts were of early
B cell lineage, with only 3% CD3-positive cells
in the diagnostic marrow. Treatment according to
Children's Cancer Group protocol
1881, regimen A, was completed in November
1994. No cranial radiation was given.
Subsequent off-therapy bone marrow aspiration
and cerebrospinal fluid (CSF) examinations were
normal.
The onset of bilateral otitis media and
pansinusitis was noted on January
30, 1995. At that time, the patient
was febrile to 38°C, the spleen was
palpable to 2 cm below the costal margin,
and right cervical adenopathy was noted. A chest
radiograph revealed diffuse bilateral
interstitial pulmonary infiltrates. Complete
blood count results were as follows: hemoglobin,
14.2 g/dL; platelets,
274 ? 109/L; white blood cells,
4.5 ? 109/L, with 13% basophils and no
blasts. The patient improved on therapy with
cefotaxime, and her splenomegaly resolved. On
February 22, 1995, she experienced a
partial complex seizure. A computed tomography
(CT) scan of the head and CSF analysis were
normal. The pulmonary infiltrates had become
nodular, and a CT-guided needle biopsy of a
nodule was obtained on March
12, 1995. The biopsy specimen was sent
for consultation, and the diagnosis of cellular
interstitial pneumonitis with features of
lymphocytic interstitial pneumonitis was
made.
By March 24, 1995, the patient had
developed marked splenomegaly and additional
seizures. Bone marrow and CSF were obtained; no
evidence of leukemia or infiltrative process was
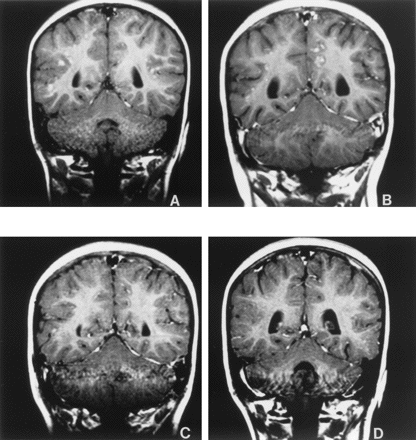
noted. An MRI scan of the head revealed multiple
gadolinium-avid cerebral nodules at the junction
of the cerebral gray and white matter in a
general distribution (Fig 1). Given the
persistence of the nodular pulmonary
infiltrates, emperic itraconazole was started.
Over the next several weeks, continued
improvement and resolution of the pulmonary
infiltrates was noted (Fig 2). However, on June
5, 1995, the patient acutely developed
a tonic pupil on the left (dilated pupil and
slow reaction to light and darkness, with
photophobia), followed 3 days later by a
decreased level of consciousness, bulbar speech,
and drooling. Complete blood count revealed:
hemoglobin, 10.9 g/dL; platelets,
168 ? 109/L; and white blood cells,
2.5 ? 109/L (46% neutrophils, 44%
lymphocytes, 10% monocytes). CSF revealed:
protein, 79 mg/dL; glucose, 50 mg/dL;
red blood cells, 1/mm3; and white blood cells,
7/mm3 (100% lymphocytes). Dexamethasone
(50 mg/m2/day divided into 6-hour
intervals) was administered, with improvement of
neurologic symptoms. The dexamethasone was then
quickly tapered to 10 mg/m2/day. On June
9, 1995, a fine-needle aspiration of
the spleen was obtained that was
nondiagnostic.
Four stereotactic needle biopsies of a brain
nodule conducted on June
13, 1995 showed an atypical lymphoid
infiltrate (Fig 3). The majority of lymphocytes
were positive for CD3 with virtually no cells
positive for L26 (CD20). Strong positivity for
CD68 was present within macrophages. The biopsy
specimens were referred for outside consultation
yielding a diagnosis of severe chronic
inflammatory changes focally consistent with
granuloma. Additional stains for acid-fast
organisms and toxoplasmosis were negative.
Ultrastructural findings showed no evidence of
significant demyelination or viral infection.
Full clinical recovery was noted by June 17,
1995, and dexamethasone was tapered.
On July 18, 1995, the patient
redeveloped splenomegaly and fever. Neck pain,
hesitant speech, and
yawning
followed, and she was again treated with
dexamethasone. This was slowly tapered, but on
August 22, 1995, she suffered an acute
decompensation with hypoxemia, tachypnea, poor
color, splenomegaly, diffuse rales, and cardiac
gallop.
An echocardiogram revealed poor cardiac
function with a shortening fraction of 20%. An
MRI of the head revealed new frontal and
parietal lesions, similar in character to those
previously seen. A CT of the abdomen revealed
new wedge-shaped densities in the kidneys,
consistent with vascular occlusion. The
following day an open-lung biopsy was obtained,
which demonstrated an atypical angiocentric
lymphoproliferative process, suggestive of
lymphomatoid granulomatosis (Fig 4).
The perivascular atypical lymphocyte
population was positive for CD45, CD3, and CD20,
the majority being CD3-positive. Pathology
consultation confirmed the immunohistologic
diagnosis of lymphomatoid granulomatosis.
Cultures of lung tissue were negative for
routine bacterial and acid-fast organisms,
fungi, and viruses. Polymerase chain reaction
analysis of frozen lung biopsy tissue showed no
clonal rearrangement of the immunoglobulin heavy
chain (IgHJH), T cell receptor -chain, and T
cell receptor -chain genes. Serology for
Epstein-Barr virus (EBV) was negative.
Serologies and cultures for cytomegalovirus were
indicative of past infection, with no evidence
of current activation. Serologies for
histoplasmosis, cryptococcus, and blastomyces
were likewise negative. EBV in situ
hybridization analysis, conducted on tissue from
the lung biopsy with intact RNA, was
negative.
Lymphoma-directed chemotherapy was initiated
on August 31, 1995 and consisted of
intravenous cyclophosphamide, intravenous
vincristine, oral prednisone, and intravenous
methotrexate.5 The patient exhibited immediate
and continued clinical improvement, and by
January 16, 1996, complete resolution
of nodules in the cerebrum and chest was noted.
Cardiac function returned to normal. The spleen,
still palpable, was markedly diminished in size.
Chemotherapy ended on March
20, 1996, by which time the
splenomegaly had resolved.
Neuropsychological testing was performed in
February 1997, 11 months after
completion of treatment for lymphomatoid
granulomatosis. Testing revealed a decrease in
overall intelligence quotient scores compared
with baseline testing completed during treatment
for her ALL in 1992. Follow-up
neuropsychological testing was administered in
March 1998, showing an improvement in
attention, fine motor speed, and coordination,
ALThough still showing deficits compared with
same-aged peers.
At her last medical follow-up in August
2000, the patient continued to be in good
health. She had been off anticonvulsants for
41 months. She attends school in a
mainstream class and receives special education
services as needed under the "other health
impaired" classification. Stimulant medication
(methylphenidate) for attention problems has
been beneficial.
DISCUSSION
The initial published series of
40 patients with lymphomatoid
granulomatosis by Liebow et al1 included only
1 child, aged 8.5 years. A subsequent
series added 116 cases, with 12 of
152 patients (8%) <20 years old.6
ALThough the primary pulmonary manifestations of
this disorder are central to the diagnosis,
extrapulmonary manifestations, as were present
in our patient's case, may be quite significant.
Involvement of the nervous system (67%), skin
(39%), kidney (32%), spleen (18%), liver (12%),
heart (11%), and lymph nodes (8%) has been
described.
The T cell origin of the disorder was first
suggested by Nichols et al and was elegantly
confirmed by Lipford and colleagues in
1988. A subsequent study of 4 patients
confirmed T cell predominance but suggested that
the process was dependent on an EBV-associated B
cell lymphoproliferative phenomenon. A more
recent series of 16 cases determined the
proliferation index of B cells, T cells, and
histiocytes in lymphomatoid granulomatosis
lesions, using combined immunohistochemistry for
CD20, CD3, CD68, and CD57 with DNA topoisomerase
II as a marker of proliferation.
The authors found a significantly higher
proliferation index in B cells compared with the
other cell populations. The average B cell
proliferation index in the high-grade (grade
III) lesions was similar to that in large cell
non-Hodgkin's B cell lymphomas. It should be
emphasized that our patient had no evidence of
EBV infection, based on serology and in situ
hybridization of pathologic lung tissue. Our
patient demonstrates that, as has also been
shown in the posttransplant lymphoproliferative
disorders, EBV need not be present to incite
this illness.
Fauci and colleagues described their
experience with 15 patients with
lymphomatoid granulomatosis, one who was
16 years old. Thirteen patients received
therapy with cyclophosphamide and prednisone;
7 had long-lasting complete remissions. Of
those who did not respond to therapy and
subsequently died, the majority developed
malignant lymphoma. Fauci et al7 noted that
early treatment with immunosuppressive therapy
markedly decreased the previously high mortality
rate (65%-90%) of lymphomatoid
granulomatosis.
We confirm that cyclophosphamide and
corticosteroid-based therapy is effective, and
note that corticosteroids alone produced only
temporary benefit in our patient. Central
nervous system benefits obtained without such
directed therapy as cranial radiation or
intrathecal chemotherapy were remarkable in this
case, and well documented by serial
neuropsychological testing and MRI.
Our patient's diagnosis was delayed in this
case because of a number of factors: 1) the
patient responded to empiric antibiotic therapy;
2) limited material was obtained from the needle
biopsies, making histopathologic review
difficult; and 3) the biopsies were all obtained
when the patient was being treated with
glucocorticoid therapy, which may have obscured
the diagnosis early in the patient's course.
Awareness of the features of this syndrome in
the appropriate clinical context may lead to
earlier recognition and prompt institution of
appropriate therapy. Childhood lymphomatoid
granulomatosis should be considered in the
clinicopathologic diagnosis of upper respiratory
tract symptomatology with concurrent nodular
pulmonary infiltrates and central nervous system
manifestations, especially in the setting of
past diagnosis of and treatment for ALL with
apparent long-term remission.