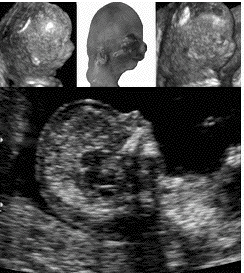
- Tous
les articles consacrés au
bâillement
foetal
- Fetal
yawning: all
publications
-
- Bien que la séquence de Pierre Robin
soit une association de malformations
oro-faciales (fente vélopalatine
postérieure, rétrognathisme,
glossoptose), elle ne constitue pas à
proprement parler une malformation fixée
de la face, dans la mesure où la plus
grande partie du phénotype va
guérir spontanément lors de la
croissance. La séquence de Pierre Robin
(communément appelée le Robin) est
plutôt une dysmorphie fonctionnelle,
c'est-à-dire une morphologie faciale
spécifique secondaire à un trouble
de la dynamique oro-faciale
anténatale.
-
- Du point de vue anténatal, la
séquence de Pierre Robin est un sujet
complexe pour plusieurs raisons. Tout d'abord
parce que les stigmates faciaux sont de
reconnaissance échographique difficile,
ensuite parce que le pronostic fonctionnel
pédiatrique est mal corrélé
aux données anatomiques, et enfin parce
que, bien qu'elle porte un nom propre qui
évoque l'idée d'une entité
bien différenciée, la
séquence de Pierre Robin est une
entité hétérogène,
tant du point de vue physiopathologique que
diagnostique, et donc pronostique. De fait, un
Robin peut être isolé ou
s'intégrer à un syndrome
malformatif, dans des proportions à peu
près égales.
-
- Tous ces éléments expliquent
pourquoi le diagnostic anténatal d'une
séquence de Pierre Robin n'est pas fait
dans la majorité des cas, et quand il est
suspecté, il met l'obstétricien et
l'échographiste dans l'embarras.
-
-
- Définition, physiopathologie et
aspect clinique
-
- 1. - La séquence de Pierre Robin
isolée
-
- Pierre Robin, stomatologiste français
du début du 20e siècle, a
décrit une nouvelle cause de
détresse vitale néonatale
constituée d'une glossoptose obstructive
et d'une rétromandibulie, aggravée
par des stigmates de vagotonie (1). Dans les
années 50, la triade malformative
associant une fente vélopalatine
postérieure, un rétrognathisme et
une glossoptose a été
décrite et nommée Pierre Robin
(2).
-
- La confusion dans les éléments
de cette définition persiste et accentue
le nombre de diagnostics effectivement en cause.
Néanmoins, la majorité des auteurs
s'accordent aujourd'hui pour dire que la triade
anatomique faciale doit être
présente pour que l'on puisse parler de
séquence de Pierre Robin. Le terme de
séquence est utilisé dans la
mesure où il apparaît que les 3
anomalies anatomiques sont liées par une
relation de cause à effet (3).
-
- Le développement embryonnaire de la
cavité orale passe par un stade (environ
45e jour de vie embryonnaire) où la
langue est verticale dans une cavité
commune nez-bouche. A ce stade, la fusion des
bourgeons maxillaires et des bourgeons
naso-frontaux est déjà faite, le
palais primaire est en place, mais la mandibule
est encore peu développée ; ni la
base du crâne, ni les maxillaires ne sont
ossifiés (4). Dans la semaine qui suit
(fin du 2e mois), les mouvements mandibulaires
antéro-postérieurs et
l'horizontalisation de la langue se mettent
parallèlement en place, et les processus
palatins secondaires issus du 1er arc branchial
peuvent se rejoindre et migrer vers la ligne
médiane et l'arrière pour former
le palais secondaire osseux. Cette étape
est sans doute brève (5). Les 3 muscles
du voile du palais se forment ensuite, ils sont
respectivement, d'avant en arrière, issus
des crêtes neurales des 3 premiers arcs
branchiaux.
-
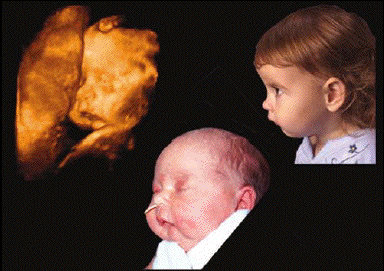
- On peut aisément concevoir qu'une
altération de cette dynamique à un
moment précis du développement
puisse être responsable d'une
séquelle anatomique, c'est-à-dire
une fente palatine postérieure, romane,
médiane, sorte d'image inversée de
la position embryonnaire de la langue. Le
mécanisme de genèse de cette fente
est fort différent de celui des fentes
labio-palatines antérieures, où
des anomalies de fusion des différents
bourgeons faciaux embryonnaires, donc une
altération des processus de morts
cellulaires peuvent être
incriminés, et ce à un stade plus
précoce du développement facial.
Cette distinction entre fente
labio-gingivo-palatine et fente palatine
postérieure de type Robin est très
importante à considérer dans
toutes les réflexions relatives aux
fentes oro-faciales du fœtus et de
l'enfant.
-
- La question est alors de savoir pourquoi la
dynamique mandibulo-linguale ne s'est pas (ou
mal) mise en place. Les causes de ce trouble de
la dynamique mandibulo-linguale sont
certainement multiples, surtout dans les Robins
intégrés à un ensemble
malformatif (6).
-
- Dans les Robins isolés, où les
enfants naissent et évoluent sans
anomalies neuromusculaires, sans autres
anomalies squelettiques, ni anomalies du
développement psychomoteur (à
condition d'une bonne prise en charge
pédiatrique), peu d'hypothèses
sont relevables. L'hypothèse d'une
compression mécanique mandibulaire
extrinsèque est théoriquement
possible, et elle est évoquée de
surcroît par la constatation
inexpliquée d'une proportion de
grossesses gémellaires plus importante
(15 %) que ne le voudrait le hasard
(fréquence des grossesses dans la
population générale = 2 %) dans
notre série de Robins isolés (n =
59).
-
- Cependant, ces Robins jumeaux n'avaient pas
d'autres signes anatomiques de contrainte
extrinsèque anténatale. De plus,
la mandibule n'est pas une partie
périphérique exposée de
l'embryon susceptible d'être l'objet d'une
contrainte mécanique.
-
- L'hypothèse la plus solide est donc
celle d'une anomalie de la mise en place ou du
contrôle de l'activité
branchiomotrice embryonnaire issue du cerveau
postérieur. Chez l'embryon humain, les
premiers mouvements observés sont des
mouvements de flexion-extension de l'axe
rachidien et des mouvements d'ouverture buccale.
Ces mouvements sont secondaires à
l'activité rythmique spontanée des
nerfs crâniens et des racines motrices
rachidiennes mises en évidence par
enregistrement de ces motoneurones du tronc
cérébral et de la moelle
isolés in vitro.
-
- Chez la souris, l'activité en
bouffées des racines motrices des nerfs
crâniens est identifiable avant la fin de
la segmentation, soit à un stade
très précoce de la vie
anténatale, avant la formation
complète du palais et l'ossification
mandibulaire (7). Cette précession du
développement des structures nerveuses
sur les structures tissulaires osseuses a
été également
montrée par des analyses histologiques
chez le fœtus humain (8). Chez la souris,
la mise en place du contrôle des fonctions
vitales (respiratoires et masticatoires) par le
tronc cérébral passe par des
stades de développement successifs
précis et très différents
d'une étape à l'autre.
-
- Au cours de ces étapes, qui
s'enchaînent de la fin de la segmentation
à la naissance, les différents
niveaux rostro-caudaux du tronc
cérébral ont des rôles
spécifiques de générateurs
de rythme et de régulateurs de la
fréquence des bouffées
d'activité des nerfs crâniens
(données personnelles). Les relations
entre ce développement fonctionnel et
l'expression des gènes de segmentation
sont en cours d'étude.
-
- Les résultats préliminaires
font apparaître un rôle du
gène krox-20 initialement connu pour
contrôler la mise en place anatomique des
rhombomères 3 et 5. L'analyse in vitro de
l'activité du réseau neuronal du
tronc cérébral des embryons de
souris krox-20 -/- montre des anomalies du
développement du rythme branchiomoteur.
De plus, l'analyse du phénotype
respiratoire et masticatoire des souris krox-20
-/- à la naissance montre de plus des
anomalies (9). Ces données montrent le
lien entre la motricité des nerfs
crâniens pendant la vie anténatale,
la morphologie oro-faciale du fœtus et la
régulation des fonctions vitales
contrôlées par le tronc
cérébral après la
naissance.
-
- De fait, les enfants atteints de syndromes
de Robin isolés présentent
à la naissance des anomalies
fonctionnelles d'intensité variable,
touchant la coordination de la
succion-déglutition, la motricité
oro-œsophagienne, la motricité
basilinguale et pharyngo-laryngée, et la
régulation ortho/parasympathique
cardiaque, qui sont autant de fonctions
normalement intégrées et
coordonnées dans la
réticulé du tronc
cérébral (10).
-
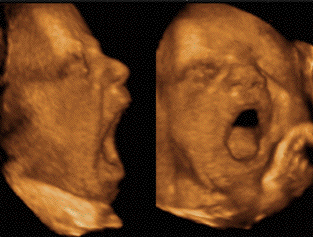
- Ces anomalies fonctionnelles ne sont pas
simplement dues au statut anatomique facial des
enfants et témoignent de la
pérennisation postnatale des anomalies
centrales responsables de la genèse de
leur dysmorphie.
-
- Ces anomalies fonctionnelles font toute la
gravité du syndrome. Elles justifient des
explorations fines : polysomnographie,
laryngoscopie dynamique, étude clinique,
vidéoscopique et/ou manométrique
de la déglutition, manométrie
œsophagienne, ECG de 24 heures avec
stimulation oculaire vagale, afin
d'évaluer la gravité de l'atteinte
du tronc cérébral et prendre les
mesures thérapeutiques adaptées
à ces défauts.
-
- La prise en charge thérapeutique de
ces enfants peut être lourde : exclusion
alimentaire initiale, nutrition entérale,
oxygénation, intubation
nasopharyngée ou trachéotomie,
éducation de l'oralité, mais elle
a permis d'obtenir de bons résultats
à long terme.
-
- La mortalité est quasi nulle (1 cas
il y a plus de 10 ans, plus depuis). La
population de Robins isolés ainsi prise
en charge va bien, tant sur le plan intellectuel
que staturopondéral et fonctionnel. Des
difficultés phonatoires et ORL justifient
un suivi et une rééducation
adaptée.
-
- 2. - Les séquences de Pierre Robin
associées à d'autres
malformations
-
- Selon les séries et les biais de
recrutement liés aux
sur-spécialités des auteurs, la
proportion d'enfants atteints de Robin avec
d'autres malformations varie, mais en moyenne
représente la moitié de l'ensemble
des cas. Les diagnostics peuvent être
divisés en diagnostics établis, on
parle alors de Robin syndromique, ou non
établis, on parle alors de Robin
associé. Cette classification a
l'intérêt de permettre de
préciser de quoi on parle. Dans notre
série de 110 Robins suivis depuis 10 ans,
56 ont une malformation associée, et pour
68 % d'entre eux, le diagnostic syndromique a
été précisé. Cinq
diagnostics dominent ce groupe de 56 enfants
(tableau I). Par ordre de fréquence, on
trouve :
-
-
- Diagnostics
- Nombre (%)
-
- Sd de Stickler et collagénopathies 15
(39 %)
- Anomalies chromosomiques 8 (21 %)
- Sd tératogénique (alcool,
antiépileptiques) 4 (10 %)
- Sd de Franceschetti 3 (7 %)
- Microdélétion 22q1.1 3 (7
%)
- Sd oculo-auriculo-vertébral 2
- Sd de Nager 1
- Sd de Kabuki 1
- Atélostéogenèse de type
I 1
- Tableau I : Diagnostics des Robins
syndromiques de notre série de 38
enfants, par ordre de fréquence.
-
- Les collagénopathies (syndrome de
Strickler et apparentés), responsables,
à des degrés variables,
d'anomalies osseuses, d'une morphologie faciale
particulière, de myopie précoce et
de déficit auditif. Ces syndromes sont
transmis sur le mode dominant et ont un
pronostic intellectuel normal.
-
- Les anomalies chromosomiques, toutes
différentes, touchant toutes des
chromosomes différents, et toutes
responsables d'atteinte psychomotrice
sévère. Les syndromes
tératogéniques, là encore
de médiocre pronostic intellectuel.
-
- Les syndromes de Franceschetti, auxquels on
peut associer le syndrome de Nager, qui sont des
malformations du 1er arc branchial, de pronostic
intellectuel satisfaisant une fois pris en
considération le risque de
surdité.
-
- Les microdélétions du
chromosome 22 (3e et 4e arc), avec des
difficultés de développement
variables. Sans aller plus loin dans cette
liste, on voit
l'hétérogénéité
majeure de ces syndromes du point de vue de leur
mécanisme (chromosome, gène,
toxique…). Les Robins associés (n =
18) ont des associations malformatives touchant
principalement le système nerveux central
et le cœur (tableau II), leur pronostic
intellectuel est en règle
médiocre.
-
- Cœur 9
- Système nerveux central 8
- Œil 6
- Squelette 5
- Autres 11
- Tableau II : Principaux organes atteints
dans les malformations des 18 Robins
associés de notre série, par ordre
de fréquence.
-
- On peut également envisager ces
Robins non isolés du point de vue de
leurs mécanismes physiopathologiques. Il
y a ceux pour lesquels l'ensemble malformatif
renforce l'idée d'une atteinte
anténatale du tronc
cérébral, comme les anomalies des
arcs branchiaux et du conotroncus cardiaque,
quelle que soit leur identité. Pour
ceux-là, une anomalie du tube neural
rhombencéphalique et des crêtes
neurales correspondante est
invoquée.
-
- Il y a ceux, à l'inverse, qui
évoquent un problème de
mobilité fœtale : syndrome
arthrogryposique, anomalies squelettiques type
pied et main bots, anomalies neuro-musculaires
périphériques. Ils sont rares.
Pour les autres, les hypothèses
physiopathologiques sont controversées,
notamment pour les Stickler, où une
anomalie primitivement osseuse est
invoquée par certains, ce qui n'est pas
notre opinion.
-
- 3. - Anatomie faciale et Pierre Robin
-
- Evaluation post-natale
-
- Plusieurs auteurs ont cherché
à quantifier les anomalies anatomiques
faciales des enfants atteints de Robin. Deux
types de travaux ont été
effectués, ceux qui cherchent à
caractériser les paramètres
faciaux des Robins à la période
néonatale, et ceux qui analysent
l'évolution de la croissance faciale des
Robins. Les méthodes de mesures sont soit
cliniques, soit radiologiques. La mesure
clinique la plus utilisée est le jaw
index qui mesure 3 paramètres :
-
- La différence
antéro-postérieure entre les 2
crêtes alvéolaires.
-
- La longueur du maxillaire supérieur,
d'une oreille à l'autre en passant par le
milieu du philtrum.
-
- La longueur de la mandibule en partant des
mêmes points auriculaires et en passant
par le milieu du menton. Le jaw index est le
résultat du calcul suivant : 1 x
2/3.
-
- Chez les Robins étudiés, le
jaw index était à 15,3 +/- 1 pour
un index à 4,2 +/- 1,8 chez les enfants
du groupe contrôle, soit un chiffre 3,6
fois supérieur chez les Robins (11). La
mesure du jaw index a l'avantage de n'être
pas invasive, et pourrait avoir un
intérêt dans l'évaluation
des rétrognathismes en
général. Les mesures radiologiques
(céphalométrie) impliquent des
tracés d'angles entre des points
précis du squelette facial et de la base
du crâne (12). Ces techniques ont toutes
une validité discutable, compte tenu des
recouvrements entre le normal et le pathologique
et compte tenu des difficultés à
valider des mesures sur une structure
circonférentielle en croissance, dont le
point de référence, le fond de la
selle turcique, bouge avec le temps.
-
- Les résultats ne sont pas fascinants
et superposables d'une méthode à
l'autre. Ils montrent que les Robins ont un
rétrognathisme significativement plus
marqué que les sujets normaux, que leur
récupération avec l'âge est
complète pour certains auteurs, partielle
pour d'autres. Une étude a montré
que les sujets avec del 22q1.1 ont des
rétrognathismes plus francs quand ils ont
un Robin associé, alors que les Stickler
ont le même morphotype facial, qu'ils
aient ou non un Robin associé. Cela peut
avoir un certain intérêt dans la
discussion physiopathologique du Robin dans ces
syndromes (13).
-
- Cependant, ces études n'ont pas un
grand intérêt pratique dans la
mesure où les pédiatres savent
qu'il y a une mauvaise corrélation entre
les stigmates anatomiques de Robin et la
gravité de leurs difficultés
fonctionnelles.
-
- De fait, dans notre série de 59
Robins isolés, nous avons
évalué les stigmates anatomiques
cliniquement (en les classant en 3 stades
d'intensité croissante), et nous avons
montré que les formes de gravité
moyenne, tant du point de vue de la taille de la
fente que de celui de l'intensité de la
glossoptose et du rétrognathisme,
dominaient en fréquence (78 % de fente
partielle, 61 % de glossoptose
modérée, 56 % de
rétrognathisme modéré).
Nous n'avons trouvé aucune
corrélation entre la taille de la fente
et la gravité fonctionnelle orodigestive
et cardiorespiratoire, et nous n'avons
trouvé de corrélation qu'entre les
formes extrêmes de rétrognathisme
et de glossoptose et les stades de
gravité fonctionnelle. Cela signifie
simplement qu'une glossoptose et un
rétrognathisme majeurs aggravent les
difficultés alimentaires et respiratoires
des enfants, ce qui n'est pas une grande
surprise.
-
- L'analyse de la face d'un enfant atteint de
Robin peut être utile au diagnostic
syndromique. Beaucoup de ces syndromes,
anomalies des arcs branchiaux bien sûr
(Franceschetti et Nager), mais également
syndrome de Stickler et syndrome
microdélétionnel 22 q11 entre
autres ont des caractères morphologiques
faciaux codifiés qui aident à la
détermination du diagnostic, surtout pour
ceux qui n'ont pas de marqueurs
moléculaires ni
cytogénétiques.
-
- 2. - Evaluation
anténatale
-
- Le diagnostic anténatal de Robin est
difficile. De fait, sur notre série de
110 cas, seulement 8 cas (7 %) avaient
été suspectés à
l'échographie anténatale, et dans
un cas au moins il s'agissait d'une
récidive dans une fratrie (tableau
III).
- Tableau III : Description des signes
échographiques dans les 8 cas de
suspicion anténatale de Robins dans notre
série de 110 cas, tous diagnostics
confondus.
-
- La fente palatine n'a été vue
qu'une seule fois, précisément
dans ce cas familial, dans la mesure où
la forme anatomique était assez grave
pour que la pointe de la langue apparaisse
au-dessus du plan du palais, ce qui n'est
possible que s'il y a une fente
postérieure. La glossoptose se traduit
par une verticalisation de la langue. Or la
langue n'est pas toujours spécifiquement
regardée et sa verticalisation peut
être modérée. De même,
le rétrognathisme ne se voit que si les
conditions échographiques sont favorables
à sa recherche (profil vrai), et s'il est
majeur.
-
- Des signes indirects, en revanche, peuvent
faire suspecter un Robin : un hydramnios et une
petite taille de l'estomac, qui sont deux signes
de réduction du débit de
déglutition du liquide amniotique.
-
- Dans notre série, l'hydramnios a
toujours été modéré,
et de ce fait non spécifique, car de tels
hydramnios sont fréquemment
rencontrés au cours de grossesses
normales, notamment chez les gros fœtus. La
petite taille de l'estomac a été
remarquée deux fois, et dans un cas, il
s'agissait d'une authentique microgastrie. De
plus, dans cette série, un Robin n'a pas
été reconnu, malgré la
compétence de l'opérateur, dans
une famille de Stickler où le risque
était précisé (50 %). A la
naissance, l'enfant avait une fente palatine
incomplète, une glossoptose et un
rétrognathisme mineurs, le diagnostic
anténatal n'était pas possible
à faire.
-
- Dans ce cas, comme dans tous probablement,
l'analyse des flux en Doppler couleur n'a pas
montré d'anomalie. Cette technique,
valide pour les fentes labio-gengivo-palatines,
ne l'est pas pour les fentes postérieures
de Robin.
-
- L'analyse du débit de
déglutition fœtale pourrait
être un élément important,
mais le débit de déglutition est
encore faible à 20-22 SA, et son
évaluation plus tardive est rendue
difficile par la position du fœtus qui
cache plus souvent son visage. De plus, sa
quantification chez le fœtus normal n'est
pas établie.
-
- Pour améliorer la performance des
diagnostics anténatals de Robins, il
faudrait que les rétrognathismes soient
dépistés par la visualisation d'un
vrai profil fœtal, que la position de la
langue (verticale ou horizontale) soit
recherchée, que la position de sa pointe
soit évaluée par rapport au plan
du palais, que sa mobilité
antéro-postérieure soit
vérifiée. Tous ces signes
devraient a fortiori être analysés
devant un hydramnios modéré ou un
petit estomac. La suspicion anténatale de
Robin doit essentiellement conduire à la
recherche de malformations associées :
face, oreille, axe des fentes
palpébrales, cardiopathie, cerveau, pieds
et mains, squelette, mobilité globale du
fœtus. Elle peut également conduire
à la réalisation d'un caryotype et
d'une analyse familiale.
-
- Le diagnostic anténatal de Robin
mène à une discussion
éthique délicate. Les anomalies
chromosomiques et les polymalformations non
étiquetées sont le plus souvent de
mauvais pronostic développemental et
justifient, à mon sens, une discussion
d'interruption thérapeutique de
grossesse. Ce n'est pas du tout le cas des
Robins isolés, même de formes
initialement graves, ni de plusieurs des Robins
syndromiques. Il ne faudrait pas que
l'amélioration des techniques
iconographiques anténatales de la face de
l'enfant (reconstruction 3D) exposent à
des risques de décisions excessives de
non poursuite de la grossesse.
- Robin P. La chute de la base de la langue
considérée comme une nouvelle
cause de gêne dans la respiration
nasopharyngienne. Bull. Acad. Med., 1923 ; 89 ;
37.
- Gorlin R.S., Cohen M.M., Levin L.S.
Syndromes of the head and the neck. 3th Ed
Oxford : Oxford University Press, 1990 ;
649-68.
- Shprintzen R.J. The implications of the
diagnosis of Robin sequence. Cleft Palate
Craniofac. J., 1992 ; 29 : 205-9.
- Couly
G. Embryogenèse normale et
pathologique de la face et du cou. Rev.
Pédiatr., 1991 ; 27 : 143-65.
- Kjaer I. Human prenatal palatal closure
related to skeletal maturity of the jaws. J.
Craniofac. Genet. Dev. Biol., 1989 ; 9 :
265-70.
- Sadewitz V.L. Robin sequence : changes in
thinking leading to changes in patient care.
Cleft Palate Craniofac. J., 1992 ; 29 :
246-52.
- Abadie V., Champagnat J., Fortin G.
Branchiomotor activities in mouse embryo.
Neuroreport, 2000 ; 11 : 141-5.
- Kjaer I. Correlated appearance of
ossification and nerve tissue in human fetal
jaws. J. Craniofac. Genet. Dev. Biol., 1990 ; 10
: 329-36.
- Jacquin T.D., Borday V., Schneider-Maunoury
S. et al. Reorganization of pontine rhythmogenic
neuronal network in Krox-20 knockout mice.
Neuron., 1996 ; 17 : 747-58.
- Abadie V., Cheron G., Couly G. Le syndrome
néonatal de dysfonctionnement du tronc
cérébral. Arch. Fr.
Pédiatr., 1993 ; 50 : 347-52.
- Van den Haven I., Mulder J.W., Van der Wal
K., Hage J.J., de Lange-de Klerk E., Haumann
T.J. The jaw index : new guide defining
micrognathia in newborns. Cleft Palate
Craniofac. J., 1997 ; 34 : 240-1.
- Laitinen S.H., Ranta R.E. Cephalometric
measurements in patients with Pierre Robin
syndrome and isolated cleft palate. Scand.
Plast. Reconstr. Hand. Surg., 1992 ; 26 :
177-83.
- Glander K., Cisneros G.J. Comparison of the
craniofacial characteristics of two syndromes
associated with the Pierre Robin sequence. Cleft
Palate Craniofac. J., 1992 ; 29 : 210-9.
|